
प्रोटीन, जैविक स्थूलअणुओं के एक महत्वपूर्ण वर्ग हैं जो सभी जैविक अवयवों में मौजूद होते हैं और मुख्य रूप से कार्बन, हाइड्रोजन, नाइट्रोजन, आक्सीजन और सल्फर तत्वों से बने होते हैं। सभी प्रोटीन, अमीनो एसिड के बहुलक हैं। अपने भौतिक आकार द्वारा वर्गीकृत किए जाने वाले प्रोटीन, नैनोकण हैं (परिभाषा: 1-100 nm). प्रत्येक प्रोटीन बहुलक - जिसे पॉलीपेप्टाइड के रूप में भी जाना जाता है - 20 अलग-अलग L-α अमीनो एसिड के अनुक्रम से बने होते हैं, जिन्हें अवशेष के रूप में भी उद्धृत किया जाता है। 40 अवशेषों के अंतर्गत श्रृंखला के लिए प्रोटीन के बजाय अक्सर पेप्टाइड शब्द का प्रयोग किया जाता है। अपने जैविक कार्यों को संपादित करने में सक्षम होने के लिए, प्रोटीन, एक या एक से अधिक विशिष्ट स्थानिक रचना में बिखर जाते हैं, जो कई गैर-सहसंयोजक अंतर्क्रिया द्वारा संचालित होते हैं जैसे हाइड्रोजन बॉन्डिंग, आयनिक अंतर्क्रिया, वान डेर वाल्स बल और जलभीतिक पैकिंग. एक आणविक स्तर पर प्रोटीन के कार्यों को समझने के लिए, उनकी त्रि-आयामी संरचना को निर्धारित करना अक्सर आवश्यक होता है। यह संरचनात्मक जीव-विज्ञान के वैज्ञानिक क्षेत्र का विषय है, जो प्रोटीन की संरचना को निर्धारित करने के लिए एक्स-रे क्रिस्टलोग्राफी, NMR स्पेक्ट्रोस्कोपी और दोहरा ध्रुवीकरण व्यतिकरण-मापी जैसी तकनीकों को लागू करता है।
एक विशेष जैव रासायनिक क्रिया संपादित करने के लिए अवशेष की कुछ विशिष्ट संख्या की आवश्यक होती है और करीब 40-50 अवशेष, कार्यात्मक डोमेन आकार के लिए निचली सीमा प्रतीत होते हैं। प्रोटीन आकार, इस निम्न सीमा से लेकर बहु-कार्यात्मक या संरचनात्मक प्रोटीन में कई हजार अवशेष तक होते हैं। हालांकि, प्रोटीन की औसत लंबाई का वर्तमान आकलन 300 अवशेष के आस-पास है।प्रोटीन उपइकाई से बहुत बड़ा समुच्चय गठित किया जा सकता है: उदाहरण के लिए, एक माइक्रोफिलामेंट में इकठ्ठा कई हजार एक्टिन अणु.g
प्रोटीन संरचना के स्तर

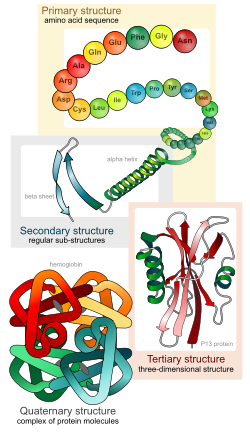
जीव रसायन-विज्ञान, एक प्रोटीन संरचना के चार अलग पहलुओं को दर्शाता है:
- प्राथमिक संरचना
- पेप्टाइड श्रृंखला का अमीनो एसिड अनुक्रम.
- माध्यमिक संरचना
- अत्यधिक नियमित उप-संरचनाएं (अल्फ़ा हेलिक्स और बीटा प्लीटेड शीट के स्ट्रैंड), जो स्थानीय रूप से परिभाषित हैं, अर्थात् एक एकल प्रोटीन अणु में कई विभिन्न माध्यमिक रूपांकन उपस्थित हो सकते हैं।
- तृतीयक संरचना
- एक एकल प्रोटीन अणु की तीन-आयामी संरचना; माध्यमिक संरचनाओं की एक स्थानिक व्यवस्था। यह, पूरी तरह से मुड़े और ठोस पॉलीपेप्टाइड श्रृंखला का भी वर्णन करता है।
- चतुर्धातुक संरचना
- प्रोटीन के कई अणुओं या पॉली पेप्टाइड श्रृंखला के संकुल, जिन्हें आम तौर पर इस संदर्भ में प्रोटीन उप-इकाई कहा जाता है, जो एक बड़े जमाव या प्रोटीन संकुल के हिस्से के रूप में कार्य करते हैं।
संरचना के इन स्तरों के अलावा, एक प्रोटीन अपनी जैविक क्रियाओं को संपादित करते हुए कई समान संरचनाओं के बीच बदलाव कर सकता है। यह प्रक्रिया पलट भी सकती है। इन कार्यात्मक पुनर्व्यवस्था के संदर्भ में ये तृतीयक या चतुर्धातुक संरचना को आम तौर पर रासायनिक रचना के रूप में संदर्भित किया जाता है और उनके बीच संक्रमण को रचनात्मक परिवर्तन कहा जाता है।
प्राथमिक संरचना, कोवैलेन्ट या पेप्टाइड बॉन्ड द्वारा बंधी रहती है, जिनका निर्माण प्रोटीन बायोसिन्थेसिस या उद्ग्रहण की प्रक्रिया के दौरान होता है। ये पेप्टाइड बॉन्ड, प्रोटीन को कठोरता प्रदान करते हैं। अमीनो एसिड श्रृंखला के दो छोर को C-टर्मिनल छोर या कार्बोज़ाइल टर्मिनस (C-टर्मिनस) कहा जाता है और N-टर्मिनल छोर या अमीनो टर्मिनस (N-टर्मिनस), प्रत्येक छोर पर मुक्त समूह की प्रकृति के आधार पर.
माध्यमिक संरचना के विभिन्न प्रकार, मुख्य श्रृंखला के पेप्टाइड समूहों के बीच हाइड्रोजन बॉन्ड की पद्धति द्वारा परिभाषित होते हैं। हालांकि, ये हाइड्रोजन बॉन्ड आम तौर पर खुद में स्थिर नहीं होते हैं, क्योंकि जल-तिक्तेय हाइड्रोजन बॉन्ड, आम तौर पर तिक्तेय-तिक्तेय हाइड्रोजन बॉन्ड की तुलना में अधिक अनुकूल होते हैं। इस प्रकार, माध्यमिक संरचना सिर्फ तभी स्थिर होती है जब जल की स्थानीय संकेंद्रता पर्याप्त रूप से न्यून होती है, जैसे, पिघली हुई गोलिका में या पूरी तरह दोहरी स्थिति में.
इसी तरह, पिघली हुई गोलिका का गठन और तृतीयक संरचना, मुख्य रूप से संरचनात्मक आधार पर गैर-विशिष्ट अंतर्क्रिया द्वारा प्रेरित होती है, जैसे अमीनो एसिड और जलभीतिक अंतर्क्रिया की रूखी प्रवृत्ति. हालांकि, तृतीयक संरचना सिर्फ तभी निश्चित होती है जब एक प्रोटीन डोमेन के हिस्से संरचनात्मक आधार पर विशिष्ट अंतर्क्रिया द्वारा जगह पर बंद कर दिए जाते हैं, जैसे आयनिक अंतर्क्रिया (लवण सेतु), हाइड्रोजन बॉन्ड और पक्ष श्रृंखला की तंग पैकिंग. बाह्य-कोशीय प्रोटीन की तृतीयक संरचना को भी डीसल्फाइड बॉन्ड द्वारा स्थिर किया जा सकता है, जो बिना दोहराव की स्थिति की एन्ट्रोपी को कम कर देता है; डीसल्फाइड बॉन्ड, साइटोसोलिक प्रोटीन में अत्यंत दुर्लभ हैं, क्योंकि साइटोसोल, आम तौर पर एक न्यूनीकरण माहौल है।
प्राथमिक संरचना
भिन्न अमीनो एसिड का अनुक्रम, प्रोटीन या पेप्टाइड की प्राथमिक संरचना कहलाता है। अवशेषों की गिनती हमेशा N-टर्मिनल छोर पर शुरू होती है (NH2 समूह), वह छोर, जहां अमीनो समूह, पेप्टाइड बॉन्ड में शामिल है। एक प्रोटीन की प्राथमिक संरचना, उस जीन से निर्धारित होती है जो उस प्रोटीन के साथ मेल खाता है। DNA में न्युक्लियोटाइड का एक विशिष्ट अनुक्रम mRNA में प्रतिरूपित होता है, जिसे रूपांतरण कही जाने वाली एक प्रक्रिया में राइबोज़ोम द्वारा पढ़ा जाता है। एक प्रोटीन का अनुक्रम उस प्रोटीन के लिए अनोखा होता है और उस प्रोटीन की संरचना और क्रिया को परिभाषित करता है। एक प्रोटीन के अनुक्रम को एडमन डीग्रडेशन या टेन्डम मास स्पेक्ट्रोमेट्री जैसे तरीकों द्वारा निर्धारित किया जा सकता है। लेकिन अक्सर, इसे उस जीन के अनुक्रम से सीधे पढ़ा जाता है जो आनुवंशिक कोड का उपयोग करता है। रूपांतरण-पश्चात के संशोधन, जैसे डीसल्फाइड निर्माण, फोस्फोरिलेशन और ग्लाइकोजाईलेशन को भी आम तौर पर प्राथमिक संरचना का एक हिस्सा माना जाता है और उसे जीन से नहीं पढ़ा जा सकता है।
माध्यमिक संरचना


बॉन्ड की लंबाई और कोण जैसी ज्ञात जानकारी का उपयोग करते हुए पेप्टाइड्स के मॉडल के निर्माण द्वारा, माध्यमिक संरचना के प्रथम तत्वों, अल्फा हेलिक्स और बीटा शीट को लिनस पौलिंग और सहकर्मियों द्वारा 1951 में सुझाया गया था। इन दो माध्यमिक संरचना तत्वों में प्रत्येक के पास एक नियमित ज्यामिति है, जिसका अर्थ है कि वे डीहाइड्रल कोण ψ और φ के विशिष्ट मूल्य से बंधे हैं। इस प्रकार, उन्हें रामचंद्रन प्लॉट के एक विशेष क्षेत्र में पाया जा सकता है। अल्फा हेलिक्स और बीटा शीट, दोनों ही पेप्टाइड रीढ़ में सभी हाइड्रोजन बॉन्ड दाताओं और स्वीकारकर्ताओं को तृप्त करने का एक तरीका प्रदर्शित करते हैं। ये माध्यमिक संरचना तत्व, केवल पॉलीपेप्टाइड मुख्य श्रृंखला के गुणों पर निर्भर करते हैं, यह समझाते हुए कि क्यों वे सभी प्रोटीन में मौजूद होते हैं। प्रोटीन का वह हिस्सा जो नियमित माध्यमिक संरचना में नहीं है, उसे एक "गैर-नियमित संरचना" कहा जाता है (यादृच्छिक कॉयल के साथ मिश्रित नहीं किया जाना चाहिए, एक खुली पॉलीपेप्टाइड श्रृंखला जिसमें किसी भी त्रि-आयामी संरचना की कमी है). उसी हेलिक्स के कुछ और निरूपण को दाईं तरफ दिखाया गया है।
सुपरमाध्यमिक संरचना
माध्यमिक संरचना के तत्वों को आम तौर पर, विभिन्न किस्मों की गांठ और मोड़ों का उपयोग करते हुए एक ठोस आकार में मोड़ा जाता है। माध्यमिक संरचनाएं, सुपरमाध्यमिक संरचनाओं के गठन के लिए हाइड्रोजन बॉन्ड द्वारा बंध जाते हैं जैसे ग्रीक की. एक विस्तृत उदाहरण के लिए देखें सुपरमाध्यमिक संरचना. यह भी कई वैज्ञानिकों द्वारा सुझाया गया है कि माध्यमिक संरचना केवल अमीनो एसिड अनुक्रम के द्वारा ही निर्धारित होती है।
तृतीयक संरचना
माध्यमिक संरचना के तत्वों को आम तौर पर, विभिन्न किस्मों की गांठ और मोड़ों का उपयोग करते हुए एक ठोस आकार में मोड़ा जाता है। तृतीयक संरचना का निर्माण आम तौर पर जलभीतिक अवशेष के दफन द्वारा प्रेरित होता है, लेकिन अन्य अंतर्क्रिया जैसे हाइड्रोजन बॉन्डिंग, आयनिक अंतर्क्रिया और डीसल्फाइड बॉन्ड भी तृतीयक संरचना को स्थिर कर सकते हैं। तृतीयक संरचना में ऐसी सभी गैर-सहसंयोजक अंतर्क्रिया शामिल हैं जिन्हें माध्यमिक संरचना नहीं माना जाता है और जो प्रोटीन के समग्र तह को परिभाषित करता है और आम तौर पर प्रोटीन की क्रियाओं के लिए अपरिहार्य है।
चतुर्धातुक संरचना
चतुर्धातुक संरचना, पेप्टाइड बॉन्ड की कई श्रृंखलाओं के बीच अंतर्क्रिया है। व्यक्तिगत श्रृंखला, उपइकाई कहलाती हैं। व्यक्तिगत उप-इकाई आम तौर पर सहसंयोजक तरीके से नहीं जुड़ी होती है, लेकिन एक डीसल्फाइड बॉन्ड से जुड़ी हो सकती है। सभी प्रोटीन में चतुर्धातुक संरचना नहीं होती है, क्योंकि वे मोनोमर्स की तरह कार्यात्मक हो सकते हैं। चतुर्धातुक संरचना को उसी श्रेणी की अंतर्क्रिया द्वारा स्थिर किया जाता है जिससे तृतीयक संरचना को किया जाता है। दो या दो से अधिक पॉलीपेप्टाइड के संकुल (यानी कई उपइकाई) को मल्टीमर कहा जाता है। विशेष रूप से यदि इसमें दो उपइकाई है तो इसे एक डाईमर कहा जाएगा, अगर तीन उपइकाई शामिल है तो ट्राईमर और चार उपइकाई के शामिल होने पर टेट्रामर कहा जाएगा. ये उपइकाईयां एक-दूसरे से आम तौर पर समरूपता धुरी द्वारा संबंधित हैं, जैसे डाईमर में एक 2-फोल्ड धुरी. समान उपइकाइयों से बने मल्टीमर को एक "होमो-" उपसर्ग के साथ संदर्भित किया जा सकता है (उदाहरण एक होमोटेट्रामर) और जो अलग-अलग उपइकाइयों से बने होते हैं उन्हें एक "हेट्रो-" उपसर्ग के साथ संदर्भित किया जा सकता है (जैसे एक हेट्रोटेट्रामर, जैसे हीमोग्लोबिन की दो अल्फा और दो बीटा श्रृंखला).
- '''''== अमीनो एसिड की संरचना ==

एक α-एमिनो एसिड, एक ऐसे हिस्से से बना होता है जो सभी प्रकार के एमिनो एसिड में मौजूद होता है और एक पक्ष श्रृंखला से जो अवशेषों के प्रत्येक प्रकार के लिए अद्वितीय होता है। Cα परमाणु, 4 भिन्न परमाणुओं से बंधे होते हैं: एक हाइड्रोजन परमाणु (चित्र में H को छोड़ा गया है), एक एमिनो समूह नाइट्रोजन, एक कार्बोज़ाइल समूह कार्बन और इस प्रकार के अमीनो एसिड के लिए विशेष एक साइड चेन कार्बन. इस नियम का अपवाद है प्रोलाइन, जहां हाइड्रोजन परमाणु, पक्ष की श्रृंखला पर एक बॉन्ड द्वारा प्रतिस्थापित किया जाता है। क्योंकि कार्बन परमाणु चार अलग-अलग समूहों के साथ बंधा होता है जिसके साथ वह काइरल है, लेकिन समावयव में से केवल एक ही जैविक प्रोटीन में मौजूद होता है। ग्लाईसीन हालांकि, काइरल नहीं है क्योंकि इसकी पक्ष श्रृंखला एक हाइड्रोजन परमाणु है। सही L-फॉर्म के लिए एक सरल स्मरक है "CORN": जब Cα परमाणु को सामने H के साथ देखा जाता है, तो अवशेष पर दक्षिणावर्त दिशा में लिखा होता है "CO-R-N".

पक्ष श्रृंखला, α-एमिनो एसिड के रासायनिक गुण को निर्धारित करता है और 20 विभिन्न पक्ष श्रृंखला के किसी भी एक को:
स्वाभाविक रूप से मौजूद होने वाले 20 अमीनो एसिड को उनके रासायनिक गुणों के आधार पर कई समूहों में विभाजित किया जा सकता है। महत्वपूर्ण कारकों में है चार्ज, हाइड्रोफोबिसिटी/हाइड्रोफिलीसिटी, आकार और कार्यात्मक समूह. जलीय पर्यावरण के साथ विभिन्न पक्ष श्रृंखला की अंतर्क्रिया की प्रकृति, प्रोटीन संरचना को ढालने में एक प्रमुख भूमिका निभाती है। हाइड्रोफोबिक पक्ष श्रृंखला, प्रोटीन के बीच में दबी होती है, जबकि हाइड्रोफिलिक पक्ष श्रृंखला विलायक के संपर्क में रहती है।
हाइड्रोफोबिक अवशेषों के उदाहरण हैं: ल्यूसीन, आइसोल्यूसीन, फेनिलएलनिन और वालीन और एक हद तक टाइरोसीन, एलानीन और ट्रिपटोफैन. पक्ष श्रृंखला का चार्ज प्रोटीन संरचनाओं में एक महत्वपूर्ण भूमिका निभाता है, क्योंकि आयन बॉन्डिंग प्रोटीन संरचनाओं को स्थिर कर सकता है और प्रोटीन के बीच में एक गैर-युग्मित चार्ज संरचना को बाधित कर सकता है। चार्ज अवशेष, बहुत अधिक हाइड्रोफिलिक होते हैं और आम तौर पर प्रोटीन के बाहर की ओर पाए जाते हैं। धनात्मक रूप से चार्ज पक्ष श्रृंखला, लाइसीन और आर्गीनीन में पाई जाती है और कुछ मामलों में हिस्टीडीन में. ऋणात्मक चार्ज ग्लुटामेट और एस्परटेट में पाए जाते हैं। शेष अमीनो एसिड में आम तौर पर विभिन्न कार्यात्मक समूहों वाली छोटी हाइड्रोफिलिक पक्ष श्रृंखला होती है। सेरीन और थ्रेओनीन में हाइड्रोक्सील समूह होता है और एस्परगिन और ग्लुटामिन में अमाइड समूह होता है। कुछ अमीनो एसिड में विशेष गुण होते हैं, जैसे सिसटाइन, जो अन्य सिसटाइन, प्रोलाइन के साथ जो चक्रीय हैं कोवैलेन्ट डिसल्फाइड बॉन्ड बना सकता है और ग्लिसीन जो छोटा और अन्य एमिनो एसिड की तुलना में अधिक लचीला है।
पेप्टाइड बॉन्ड (अमाइड बॉन्ड)
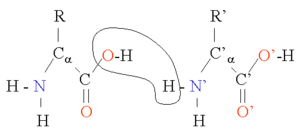

दो अमीनो एसिड को संक्षेपण अभिक्रिया में संयुक्त किया जा सकता है। इस अभिक्रिया को दोहराकर, अवशेषों की लंबी श्रृंखला (पेप्टाइड बॉन्ड में अमीनो एसिड) को उत्पन्न किया जा सकता है। यह अभिक्रिया राइबोजोम द्वारा उत्प्रेरित होती है और उस प्रक्रिया को रूपांतरण के रूप में जाना जाता है। पेप्टाइड बॉन्ड, डबल बॉन्ड से इलेक्ट्रॉन के गैर-स्थानीयकरण के कारण वास्तव में प्लानर हैं। कठोर पेप्टाइड डिहेड्राल कोण, ω (N और C1 के बीच बॉन्ड) हमेशा 180 डिग्री के नज़दीक होता है। डिहेड्राल कोण phi φ (Cα और N के बीच बॉन्ड) और psi ψ (Cα और C1 के बीच बॉन्ड) में संभावित मान की एक निश्चित श्रेणी हो सकती है। ये कोण एक प्रोटीन की स्वतंत्रता के स्तर हैं, वे प्रोटीन की तीन आयामी संरचना का नियंत्रण करते हैं। वे ज्यामिति द्वारा सीमित होते हैं ताकि विशिष्ट माध्यमिक संरचना तत्वों के लिए विशिष्ट श्रेणियों को अनुमति दे सकें और रामचंद्रन प्लॉट में प्रदर्शित होते हैं। कुछ महत्वपूर्ण बॉन्ड लंबाई नीचे तालिका में दी गई है।
| पेप्टाइड बॉन्ड | औसत लंबाई | एकल बॉन्ड | औसत लंबाई | हाइड्रोजन बॉन्ड | औसत (30 ±) |
| Ca - C | 153 pm | C - C | 154 pm | O-H --- O-H | 280 pm |
| C - N | 133 pm | C - N | 148 pm | N-H --- O=C | 290 pm |
| N - Ca | 146 pm | C - O | 143 pm | O-H --- O=C | 280 pm |
पक्ष-श्रृंखला रचना और रोटामर्स
पक्ष श्रृंखला के साथ लगे हुए परमाणुओं को ग्रीक वर्णमाला क्रम में ग्रीक अक्षरों द्वारा नाम दिया जाता है: α, β, γ, δ, є और इसी तरह. Cα रीढ़ के उस कार्बन परमाणु को संदर्भित करता है जो उस अमीनो एसिड, Cβ के कार्बोनिल समूह के निकटतम है, दूसरा सबसे निकटतम और इसी तरह आगे. Cα रीढ़ का एक हिस्सा है, जबकि Cβ और बाहर के परमाणु, पक्ष श्रृंखला का निर्माण करते हैं। इन परमाणुओं के बीच बॉन्ड के आसपास के डिहेड्रल कोण को χ1, χ2, χ3 आदि नाम दिया गया है। पक्ष श्रृंखला के प्रथम गतिशील परमाणु का डिहेड्रल कोण, , जिसे परिभाषित किया गया है N-C-C- के रूप में, उसे नाम दिया है χ1. अधिकांश पक्ष श्रृंखला, भिन्न रचना में हो सकते हैं जिसे गौश (-), ट्रांस और गौश (+) कहा जाता है। पक्ष श्रृंखला, आम तौर पर कंपित रचना में χ2 के आसपास आने का एक प्रयास करती हैं, जो प्रतिस्थापन परमाणुओं के इलेक्ट्रॉन कक्षीय के बीच ओवरलैप के न्यूनीकरण द्वारा प्रेरित होती है।




पक्ष-श्रृंखला की रचना की विविधता अक्सर रोटामर लाइब्रेरी में व्यक्त की जाती है। एक रोटामर लाइब्रेरी, पक्ष-श्रृंखला स्वतंत्रता के स्तर वाले प्रोटीन में प्रत्येक प्रकार के अवशेष के लिए रोटामर्स का संग्रह है। रोटामर लाइब्रेरीयों में आम तौर पर रचना और एक निश्चित रचना की आवृत्ति, दोनों के बारे में जानकारी होती है। लाइब्रेरियों में अक्सर डिहेड्रल कोण तरीके या पद्धति के बारे में विचरण के बारे में जानकारी शामिल होती है, जिसे नमूनों में इस्तेमाल किया जा सकता है।
पक्ष-श्रृंखला डिहेड्रल कोण, समान रूप से वितरित नहीं हैं, लेकिन अधिकांश पक्ष-श्रृंखला प्रकार के लिए, कोण, किसी निश्चित मूल्यों के आसपास और तंग समूहों में मौजूद होते हैं। रोटामर लाइब्रेरियों को इसलिए आम तौर पर प्रोटीन की ज्ञात संरचनाओं में पक्ष-श्रृंखला रचना के सांख्यिकीय विश्लेषण से निकाला जाता है जिसके लिए पाई गई रचना को इकठ्ठा किया जाता है या डिहेड्रल कोण स्थान को बिन में विभाजित करके और फिर प्रत्येक बिन में एक औसत रचना का निर्धारण करके. यह विभाजन आम तौर पर भौतिक, रासायनिक आधार पर होता है, जैसा कि SP3-SP3 बॉन्ड के रोटेशन का 120° बिन में विभाजन जो प्रत्येक कम्पित रचना पर केन्द्रित होता है (60°, 180°,-60°).

रोटामर लाइब्रेरियां रीढ़-स्वतंत्र, माध्यमिक-संरचना पर निर्भर, या रीढ़-निर्भर हो सकती हैं। भेद को इस आधार पर किया जाता है कि क्या रोटामर के लिए डिहेड्रल कोण और/या उनकी आवृत्तियां स्थानीय रीढ़ रचना पर निर्भर करती हैं या नहीं। रीढ़-स्वतंत्र रोटामर लाइब्रेरियां, रीढ़ रचना को संदर्भित नहीं करती हैं और इनकी गणना एक निश्चित प्रकार की उपलब्ध पक्ष-श्रृंखला से की जाती है। माध्यमिक-संरचना पर निर्भर लाइब्रेरियां, भिन्न डिहेड्रल कोण और/या -हेलिक्स, -शीट के लिए रोटामर आवृत्तियां या कॉयल माध्यमिक संरचनाओं को प्रस्तुत करती हैं। रीढ़-निर्भर रोटामर लाइब्रेरियां रचना और/या स्थानीय आवृत्तियों पर निर्भर रीढ़ रचना जैसा कि रीढ़ डिहेड्रल कोण और द्वारा परिभाषित है, माध्यमिक संरचना पर ध्यान दिए बिना. अंत में, रीढ़-निर्भर रोटामर लाइब्रेरी का भिन्न रूप, स्थिति विशेष रोटामर के रूप में मौजूद है, जो आम तौर पर 5 अमीनो एसिड की लंबाई वाले टुकड़े से परिभाषित होता है, जहां केंद्रीय अवशेष की पक्ष श्रृंखला रचना की जांच होती है।


प्रोटीन संरचना में डोमेन, रूपांकन और परत
कई प्रोटीन, कई इकाइयों में व्यवस्थित होते हैं। एक संरचनात्मक डोमेन, प्रोटीन की समग्र संरचना का तत्त्व है जो स्वयं-स्थिर है और शेष प्रोटीन श्रृंखला से अक्सर स्वतन्त्र रूप से मुड़ता है। कई डोमेन, एक जीन या जीन परिवार के प्रोटीन उत्पादों के लिए अनोखे नहीं होते हैं बल्कि विभिन्न प्रोटीन में दिखाई देते हैं। डोमेन को अक्सर नाम दिया जाता है और उन्हें बाहर किया जाता है क्योंकि वे उस प्रोटीन की जैविक क्रियाओं में प्रमुख रूप से उपस्थित होते हैं जिसके अंतर्गत वे आते हैं; उदाहरण के लिए "काल्मोडुलिन का कैल्शियम-बाइंडिंग डोमेन". क्योंकि वे स्वयं-स्थिर हैं, डोमेन को आनुवांशिक इंजीनियरिंग द्वारा, काइमेरा बनाने के लिए एक प्रोटीन से दूसरे प्रोटीन में "बदला" जा सकता है। इस अर्थ में एक रूपांकन, संरचनात्मक माध्यमिक तत्वों के एक छोटे विशिष्ट संयोजन को दर्शाता है (जैसे हेलिक्स-टर्न-हेलिक्स). इन तत्वों को अक्सर सुपरमाध्यमिक संरचना कहा जाता है। फोल्ड का तात्पर्य एक वैश्विक प्रकार की व्यवस्था से होता है, जैसे हेलिक्स बंडल या बीटा-बैरल. संरचना रूपांकन आम तौर पर, केवल कुछ तत्वों से मिलकर बने होते हैं, जैसे 'हेलिक्स-टर्न-हेलिक्स' में सिर्फ तीन हैं। ध्यान दें कि जबकि एक रूपांकन के सभी उदाहरणों में तत्वों के स्थानिक अनुक्रम समान हैं, अंतर्निहित जीन के भीतर उन्हें किसी भी क्रम में कूटित किया जा सकता है। संरचनात्मक प्रोटीन रूपांकनों में अक्सर चर लंबाई और अनिर्दिष्ट संरचना के लूप शामिल होते हैं, जो प्रभावस्वरूप "स्लैक" का निर्माण करता है जो उन दो तत्वों को स्थान में लाने के लिए आवश्यक है जो सबसे निकटवर्ती DNA अनुक्रम द्वारा कूटित नहीं होते हैं। यह भी ध्यान दें है कि यहां तक कि, जब दो जीन, एक ही क्रम में एक रूपांकन के माध्यमिक संरचनात्मक तत्वों को कूटित करते हैं, तब पर भी वे अमीनो एसिड के, कुछ हद तक भिन्न कनुक्रम को निर्दिष्ट कर सकते हैं। यह न केवल तृतीयक और प्राथमिक संरचना के बीच जटिल संबंधों की वजह से सच है, बल्कि इसलिए क्योंकि तत्वों का आकार एक प्रोटीन से दूसरे प्रोटीन में बदलता है। इस तथ्य के बावजूद कि युकैरीओटिक सिस्टम में करीब 100,000 प्रोटीन व्यक्त किए गए हैं, वहां अपेक्षाकृत कम भिन्न डोमेन, संरचनात्मक रूपांकन और मोड़ हैं। यह आंशिक रूप से विकास का एक परिणाम है, क्योंकि जीन और जीन के हिस्से दोगुने किए जा सकते हैं या जीनोम के भीतर चलाए जा सकते हैं। इसका मतलब है कि, उदाहरण के लिए, एक प्रोटीन डोमेन को एक प्रोटीन से दूसरे में ले जाया जा सकता है और इस प्रकार प्रोटीन को एक नया कार्य सौंपा जा सकता है। इन तंत्रों वजह से मार्गों और तंत्रों का कई अलग-अलग प्रोटीन में पुनः उपयोग किया जा सकता है।
प्रोटीन तह
एक बिना तह किया हुआ पॉलीपेप्टाइड, यादृच्छिक कॉयल से तीन-आयामी संरचना की अपनी विशेषता में फोल्ड होता है।
प्रोटीन संरचना का निर्धारण
प्रोटीन डाटा बैंक में उपलब्ध लगभग 90% प्रोटीन संरचनाओं का एक्स रे क्रिस्टलोग्राफी द्वारा निर्धारण किया गया है। यह विधि प्रोटीन (क्रिस्टल रूप में) में इलेक्ट्रॉन के 3D घनत्व वितरण को मापने की अनुमति देती है और जिससे किसी विशिष्ट रिजोल्यूशन में निर्धारित किये जाने वाले सभी परमाणुओं का 3D निर्देशांक का अनुमान लगाया जा सकता है। मोटे तौर पर, ज्ञात प्रोटीन संरचनाओं में 9% को परमाणु चुंबकीय अनुनाद तकनीक द्वारा प्राप्त किया जाता है, जिसका इस्तेमाल माध्यमिक संरचना को निर्धारित करने के लिए भी किया जा सकता है। ध्यान दें कि समग्र रूप से माध्यमिक संरचना के पहलुओं को अन्य जैव रासायनिक तकनीकों के माध्यम से निर्धारित किया जा सकता है, जैसे कि सर्कुलर डाईक्रोइज़म या डुअल पोलराईज़ेशन इंटरफेरोमेट्री. एक उच्च सटीकता के साथ माध्यमिक संरचना को पूर्वानुमानित भी किया जा सकता है (अगला अनुभाग देखें). क्रायो-इलेक्ट्रॉन माइक्रोस्कोपी, हाल के समय में उच्च रेजोल्यूशन में प्रोटीन संरचनाओं का निर्धारण करने का साधन बन गई है (5 आंग्सस्टोर्म से कम या 0.5 नैनोमीटर) और अगले दशक में एक उच्च रेजोल्यूशन के लिए एक उपकरण के रूप में इसके अधिक शक्तिशाली होने की अपेक्षा की जाती है। यह तकनीक उन शोधकर्ताओं के लिए अभी भी एक बहुमूल्य संसाधन है जो बहुत बड़े प्रोटीन संकुल के साथ काम करते हैं जैसे वायरस कोट प्रोटीन और एमीलोयड फाइबर.
| रेजोल्यूशन (A) | अर्थ |
| >4.0 | व्यक्तिगत अर्थहीन निर्देशांक |
| 3.0-4.0 | तह संभवतः सही है, लेकिन त्रुटियों की बहुत संभावना है। कई पक्ष श्रृंखला गलत रोटामर के साथ रखी हैं। |
| 2.5-3.0 | तह के सही होने की संभावना, सिवाय इसके कि सतह के कुछ लूप संभवतः गलत मॉडल के हों. कई लंबी, पतली पक्ष श्रृंखला (lys, glu, gln, आदि) और छोटी पक्ष श्रृंखला (ser, val, thr, आदि) में गलत रोटामर होने की संभावना है। |
| 2.0-2.5 | 2.5 - 3.0 के रूप में, लेकिन गलत रोटामर में पक्ष श्रृंखला की संख्या काफी कम है। कई छोटी त्रुटियों को सामान्य रूप से पाया जा सकता है। तह सामान्य रूप से सही और सतह लूप में त्रुटियों की संख्या कम. पानी के अणु और छोटे लिगेंड दिखाई देने लगते हैं। |
| 1.5-2.0 | बहुत कम अवशेष में गलत रोटामर है। कई छोटी त्रुटियों को सामान्य रूप से पाया जा सकता है। तह शायद ही कभी गलत होते हैं, यहां तक कि सतह लूप में भी नहीं। |
| 0.5-1.5 | सामान्य में, संरचनाओं में इस रेजोल्यूशन में लगभग कोई त्रुटि नहीं है। रोटामर लाइब्रेरी और ज्यामितीय अध्ययन इन संरचनाओं से बने हैं। |
संरचना वर्गीकरण
प्रोटीन संरचनाओं को उनकी समानता या आम विकासवादी मूल के आधार पर वर्गीकृत किया जा सकता है। SCOP और CATH डेटाबेस, दो भिन्न संरचनात्मक वर्गीकरण प्रदान करते हैं।
प्रोटीन संरचना का अभिकलनात्मक पूर्वानुमान
प्रोटीन अनुक्रम का निर्माण प्रोटीन संरचना के निर्माण की तुलना में बहुत सरल है। हालांकि, प्रोटीन की संरचना, उसके अनुक्रम की तुलना में प्रोटीन की क्रियाओं की अधिक जानकारी देती है। इसलिए, प्रोटीन संरचना के उसके अनुक्रम से अभिकलनात्मक पूर्वानुमान के लिए कई तरीकों को प्रस्तावित किया गया है। प्रारंभिक पूर्वानुमान तरीके प्रोटीन के बस अनुक्रम का उपयोग करते हैं। थ्रेडिंग मौजूदा प्रोटीन संरचनाओं का उपयोग करता है। ज्ञात संरचना के एक या एक से अधिक प्रोटीन से अज्ञात संरचना के एक प्रोटीन के एक विश्वसनीय 3D मॉडल बनाने के लिए अनुरूपता मॉडलिंग. प्रोटीन संरचना पूर्वानुमान में हाल की प्रगति और चुनौतियों की जांग द्वारा समीक्षा की गई.
प्रोटीन संरचना से संबंधित सॉफ्टवेयर
प्रोटीन संरचना के विभिन्न पहलुओं पर, अक्सर अतिव्यापी, काम कर रहे शोधकर्ताओं की सहायता के लिए सॉफ्टवेयर मौजूद हैं। सबसे बुनियादी कार्यशीलता, संरचना कल्पना प्रदान करती है। प्रोटीन संरचना के विश्लेषण को ऐसे सॉफ्टवेयर द्वारा सरल किया जा सकता जो संरचना को पंक्तिबद्ध करता है। दिए गए प्रोटीन अनुक्रम के लिए एक मौजूदा संरचनाओं के अभाव में, ऐसे तरीके मौजूद हैं जिनसे ज्ञात प्रोटीन संरचना पर आधारित ऐसे अनुक्रम की संरचना को पूर्वानुमानित या स्वरूपित किया जा सकता है। और ज्ञात या पूर्वानुमानित संरचनाओं के दिए गए मॉडल से कोई व्यक्ति सॉफ्टवेयर का उपयोग त्रुटियों के लिए उनको सत्यापित करने हेतु, प्रोटीन रचना परिवर्तन के पूर्वानुमान के लिए, या अधःस्तरीय बाध्यकारी साइटों के पूर्वानुमान के लिए कर सकता है।
इन्हें भी देखें
अतिरिक्त पठन
- Chiang YS, Gelfand TI, Kister AE, Gelfand IM (2007). "New classification of supersecondary structures of sandwich-like proteins uncovers strict patterns of strand assemblage". Proteins. 68 (4): 915–921. PMID 17557333. डीओआइ:10.1002/prot.21473.सीएस1 रखरखाव: एक से अधिक नाम: authors list (link)
- Habeck M, Nilges M, Rieping W (2005). "Bayesian inference applied to macromolecular structure determination" (PDF). Physical review. E, Statistical, nonlinear, and soft matter physics. 72 (3 Pt 1): 031912. PMID 16241487. मूल (PDF) से 30 सितंबर 2007 को पुरालेखित. अभिगमन तिथि 24 जून 2010.सीएस1 रखरखाव: एक से अधिक नाम: authors list (link) (NMR डाटा से संरचना निर्धारण के लिए बायेसियन अभिकलनात्मक पद्धति)
बाहरी कड़ियाँ
- SSS Database - सुपर-माध्यमिक प्रोटीन संरचना डेटाबेस
- SPROUTS (स्ट्रक्चरल प्रेडिक्शन फॉर प्रोटीन फोल्डिंग यूटिलिटी सिस्टम)
- ProSA-web - प्रयोगात्मक या सिद्धांततः निर्धारित प्रोटीन संरचनाओं में त्रुटियों की पहचान के लिए एक वेब सेवा
- NQ-Flipper - प्रोटीन संरचनाओं में Asn और Gln अवशेष के प्रतिकूल रोटामर के लिए जांच
- WHAT IF servers - प्रोटीन संरचना के करीब 200 पहलुओं की जांच करता है, जैसे पेकिंग, ज्यामिति, Asn, Gln के लिए सामान्य रूप से प्रतिकूल रोटामर की, विशेष रूप से, अजीब पानी अणु, रीढ़ रचना, एटम नामकरण, समरूपता मानक, आदि
- Bioinformatics course - एक इंटरैक्टिव, पूरी तरह से स्वतंत्र, पाठ्यक्रम जो wiki की इस प्रविष्टि में चर्चा किए गए पहलुओं की व्याख्या करता है।